thermodynamics
- the branch of physical science that deals with the relations between heat and other forms of energy (such as mechanical, electrical, or chemical energy), and, by extension, of the relationships between all forms of energy. is known as thermodynamics
Laws of thermodynamics
- Zeroth law of thermodynamics: If two systems are in thermal equilibrium respectively with a third system, they must be in thermal equilibrium with each other. This law helps define the notion of temperature
- First law of thermodynamics: When energy passes, as work, as heat, or with matter, into or out from a system, its internal energychanges in accord with the law of conservation of energy. Equivalently, perpetual motion machines of the first kind are impossible.
- Second law of thermodynamics: In a natural thermodynamic process, the sum of the entropies of the participatingthermodynamic systems increases. Equivalently, perpetual motion machines of the second kind are impossible.
- Third law of thermodynamics: The entropy of a system approaches a constant value as the temperature approaches absolute zero.[2] With the exception of glasses the entropy of a system at absolute zero is typically close to zero, and is equal to the log of the multiplicity of the quantum ground state.
Zeroth law
The law is intended to allow the existence of an empirical parameter, the temperature, as a property of a system such that systems in thermal equilibrium with each other have the same temperature. The law as stated here is compatible with the use of a particular physical body, for example a mass of gas, to match temperatures of other bodies, but does not justify regarding temperature as a quantity that can be measured on a scale of real numbers. it is only one of a diversity of statements that are labeled as "the zeroth law" by competent writers. Some statements go further so as to supply the important physical fact that temperature is one-dimensional, that one can conceptually arrange bodies in real number sequence from colder to hotter.
First law
The first law of thermodynamics is a version of the law of conservation of energy, adapted for thermodynamic systems. The law of conservation of energy states that the total energy of an isolated system is constant; energy can be transformed from one form to another, but cannot be created or destroyed. The first law is often formulated by stating that the change in the internal energy of aclosed system is equal to the amount of heat supplied to the system, minus the amount of work done by the system on its surroundings. Equivalently, perpetual motion machines of the first kind are impossible.
The first law of thermodynamics may be stated in several ways:
- The increase in internal energy of a closed system is equal to the heat supplied to the system minus work done by it.

- For a thermodynamic cycle of a closed system, which returns to its original state, the heat Qin supplied to a closed system in one stage of the cycle, minus that Qout removed from it in another stage of the cycle, equals the net work done by the system.
-
 , and, consequently
, and, consequently 
- The increase in internal energy of a closed adiabatic system can only be the result of the net work performed by the system, because Q = 0.
-

-
-
- This states that energy can be neither created nor destroyed. However, energy can change forms, and energy can flow from one place to another. The total energy of an isolated system does not change.
- The concept of internal energy and its relationship to temperature.
-
- If a system has a definite temperature, then its total energy has three distinguishable components. If the system is in motion as a whole, it has kinetic energy. If the system as a whole is in an externally imposed force field (e.g. gravity), it has potential energy relative to some reference point. Finally, it has internal energy, which is a fundamental quantity for thermodynamics. Beyond the conceptual frame of macroscopic thermodynamics, it can be explained as the sum of the disorganized kinetic energy of microscopic motions of its constituent atoms, and of the potential energy of interactions between them. Other things being equal, the kinetic energy of microscopic motions of the constituent atoms increases as the system's temperature increases. The establishment of the concept of internal energy is the characteristic distinguishing feature of the first law of thermodynamics.
- The flow of heat is a form of energy transfer.
-
- Heating is a natural process of moving energy to or from a system other than by work or the transfer of matter. The heat passes only from a hotter to a colder system.
-
-
- If the system has rigid walls impermeable to matter, and no external long-range force field affects it, and consequently energy cannot be transferred as work into or out from the system then:
-
where Q denotes the amount of energy transferred into the system as heat.- Work is a process of transferring energy to or from a system. Unless otherwise stated, it is customary to treat work as supplied without dissipation in the surroundings. Within the system, in a natural process, some of the transferred work is dissipated.
-
- For example, when a machine lifts a system upwards, some energy is transferred from the machine to the system. The system acquires its energy in the form ofgravitational potential energy in this example.
-
- Or in general it can be partitioned to kinetic, potential or internal energy
- When matter is transferred, its associated internal energy is transferred with it.
where uexternal denotes the internal energy per unit mass of the transferred matter, measured when it is still in the surroundings, before transfer; and ΔM denotes the transferred mass.Combining these principles leads to one traditional statement of the first law of thermodynamics: it is not possible to construct a machine which will perpetually output work without an equal amount of energy input to that machine. Or more briefly, a perpetual motion machine is impossible. -
Conceptually revised statement, according to the mechanical approach
- he revised statement of the law takes the notions of adiabatic mechanical work, and of non-adiabatic transfer of energy, as empirically or theoretically established primitive notions. It rests on the primitive notion of walls, especially adiabatic walls, presupposed as physically established. Energy can pass such walls as only as adiabatic work, reversibly or irreversibly. If transfer of energy as work is not permitted between them, two systems separated by an adiabatic wall can come to their respective internal mechanical and material thermodynamic equilibrium states completely independently of one another.[13]The revised statement of the law postulates that a change in the internal energy of a system due to an arbitrary process of interest, that takes the system from its specified initial to its specified final state of internal thermodynamic equilibrium, can be determined through the physical existence of a reference process, for those specified states, that occurs purely through stages of adiabatic work.The revised statement is then
-
- For a closed system, in any arbitrary process of interest that takes it from an initial to a final state of internal thermodynamic equilibrium, the change of internal energy is the same as that for a reference adiabatic work process that links those two states. This is so regardless of the path of the process of interest, and regardless of whether it is an adiabatic or a non-adiabatic process. The reference adiabatic work process may be chosen arbitrarily from amongst the class of all such processes.
This statement is much less close to the empirical basis than are the original statements,[14] but is often regarded as conceptually parsimonious in that it rests only on the concepts of adiabatic work and of non-adiabatic processes, not on the concepts of transfer of energy as heat and of empirical temperature that are presupposed by the original statements. Largely through the influence of Max Born, it is often regarded as theoretically preferable because of this conceptual parsimony. Born particularly observes that the revised approach avoids thinking in terms of what he calls the "imported engineering" concept of heat engines.[10]Basing his thinking on the mechanical approach, Born in 1921, and again in 1949, proposed to revise the definition of heat.[10][15] In particular, he referred to the work ofConstantin Carathéodory, who had in 1909 stated the first law without defining quantity of heat.[16] Born's definition was specifically for transfers of energy without transfer of matter, and it has been widely followed in textbooks (examples:[17][18][19]). Born observes that a transfer of matter between two systems is accompanied by a transfer of internal energy that cannot be resolved into heat and work components. There can be pathways to other systems, spatially separate from that of the matter transfer, that allow heat and work transfer independent of and simultaneous with the matter transfer. Energy is conserved in such transfers. -
Description
- For a closed system, in any process, the change in the internal energy is considered due to a combination of heat added to the system and work done by the system. Taking
 as a change in internal energy, one writeswhere
as a change in internal energy, one writeswhere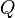 and
and  are quantities of heat supplied to the system by its surroundings and of work done by the system on its surroundings, respectively. This sign convention is implicit in Clausius' statement of the law given above, and is consistent with the use of thermodynamics to study heat engines, which provide useful work that is regarded as
are quantities of heat supplied to the system by its surroundings and of work done by the system on its surroundings, respectively. This sign convention is implicit in Clausius' statement of the law given above, and is consistent with the use of thermodynamics to study heat engines, which provide useful work that is regarded as - When a system expands in a fictive quasistatic process, the work done by the system on the environment is the product, P dV, of pressure, P, and volume change, dV, whereas the work done on the system is -P dV. Using either sign convention for work, the change in internal energy of the system is:
arious statements of the law for closed systems
- For the thermodynamics of closed systems, the distinction between transfers of energy as work and as heat is central and is within the scope of the present article. For the thermodynamics of open systems
-
-
-
-
-
- For a given system we let ΔE kin = large-scale mechanical energy, ΔE pot = large-scale potential energy, and ΔE tot = total energy. The first two quantities are specifiable in terms of appropriate mechanical variables, and by definition
-
-
-
-
-
-
-
-
-
- For any finite process, whether reversible or irreversible,
-
-
-
-
-
-
-
-
-
- The first law in a form that involves the principle of conservation of energy more generally is
-
-
-
-
-
-
-
-
-
- Here Q and W are heat and work added, with no restrictions as to whether the process is reversible, quasistatic, or irreversible
Adiabatic processes
- In an adiabatic process, there is transfer of energy as work but not as heat. For all adiabatic process that takes a system from a given initial state to a given final state, irrespective of how the work is done, the respective eventual total quantities of energy transferred as work are one and the same, determined just by the given initial and final states. The work done on the system is defined and measured by changes in mechanical or quasi-mechanical variables external to the system. Physically, adiabatic transfer of energy as work requires the existence of adiabatic enclosures.
General case for reversible processes
-
-
-
-
-
- Work transfer is practically reversible when it occurs so slowly that there are no frictional effects within the system; frictional effects outside the system should also be zero if the process is to be globally reversible. For a particular reversible process in general, the work done reversibly on the system,
 , and the heat transferred reversibly to the system,
, and the heat transferred reversibly to the system,  are not required to occur respectively adiabatically or adynamically, but they must belong to the same particular process defined by its particular reversible path,
are not required to occur respectively adiabatically or adynamically, but they must belong to the same particular process defined by its particular reversible path,  , through the space of thermodynamic states.
, through the space of thermodynamic states. - e first law for a particular reversible process can be written

- This combined statement is the expression the first law of thermodynamics for reversible processes for closed systems.In particular, if no work is done on a thermally isolated closed system we have
 .
.General case for irreversible processes
- the energy transfer is not under a practically zero temperature gradient and practically frictionless, then the process is irreversible. Then the heat and work transfers may be difficult to calculate, and irreversible thermodynamics is called for. Nevertheless, the first law still holds and provides a check on the measurements and calculations of the work done irreversibly on the system,
 , and the heat transferred irreversibly to the system,
, and the heat transferred irreversibly to the system,  , which belong to the same particular process defined by its particular irreversible path,
, which belong to the same particular process defined by its particular irreversible path, - This means that the internal energy
 is a function of state and that the internal energy change between two states is a function only of the two states.
is a function of state and that the internal energy change between two states is a function only of the two states.
First law of thermodynamics for open systems
- For the first law of thermodynamics, there is no trivial passage of physical conception from the closed system view to an open system view.[55][56] For closed systems, the concepts of an adiabatic enclosure and of an adiabatic wall are fundamental. Matter and internal energy cannot permeate or penetrate such a wall. For an open system, there is a wall that allows penetration by matter. In general, matter in diffusive motion carries with it some internal energy, and some microscopic potential energy changes accompany the motion. An open system is not adiabatically enclosed.
Second law
- The second law of thermodynamics asserts the irreversibility of natural processes, and the tendency of natural processes to lead towards spatial homogeneity of matter and energy, and especially of temperature. It can be formulated in a variety of interesting and important ways.It implies the existence of a quantity called the entropy of a thermodynamic system. In terms of this quantity it implies thatThis statement of the law recognizes that in classical thermodynamics, the entropy of a system is defined only when it has reached its own internal thermodynamic equilibrium.The second law refers to a wide variety of processes, reversible and irreversible. All natural processes are irreversible. Reversible processes are a convenient theoretical fiction and do not occur in nature.A prime example of irreversibility is in the transfer of heat by conduction or radiation. It was known long before the discovery of the notion of entropy that when two bodies initially of different temperatures come into thermal connection, then heat always flows from the hotter body to the colder one.The second law tells also about kinds of irreversibility other than heat transfer, for example those of friction and viscosity, and those of chemical reactions. The notion of entropy is needed to provide that wider scope of the law.According to the second law of thermodynamics, in a theoretical and fictional reversible heat transfer, an element of heat transferred, δQ, is the product of the temperature (T), both of the system and of the sources or destination of the heat, with the increment (dS) of the system's conjugate variable, its entropy (S)Entropy may also be viewed as a physical measure of the lack of physical information about the microscopic details of the motion and configuration of a system, when only the macroscopic states are known. The law asserts that for two given macroscopically specified states of a system, there is a quantity called the difference of information entropy between them. This information entropy difference defines how much additional microscopic physical information is needed to specify one of the macroscopically specified states, given the macroscopic specification of the other - often a conveniently chosen reference state which may be presupposed to exist rather than explicitly stated. A final condition of a natural process always contains microscopically specifiable effects which are not fully and exactly predictable from the macroscopic specification of the initial condition of the process. This is why entropy increases in natural processes - the increase tells how much extra microscopic information is needed to distinguish the final macroscopically specified state from the initial macroscopically specified state
Carnot's principle
- It refers to a cycle of a Carnot engine, fictively operated in the limiting mode of extreme slowness known as quasi-static, so that the heat and work transfers are between subsystems that are always in their own internal states of thermodynamic equilibrium. The Carnot engine is an idealized device of special interest to engineers who are concerned with the efficiency of heat engines. Carnot's principle was recognized by Carnot at a time when the caloric theory of heat was seriously considered, before the recognition of the first law of thermodynamics, and before the mathematical expression of the concept of entropy. Interpreted in the light of the first law, it is physically equivalent to the second law of thermodynamics, and remains valid today. It states
Clausius statement
- The statement by Clausius uses the concept of 'passage of heat'. As is usual in thermodynamic discussions, this means 'net transfer of energy as heat', and does not refer to contributory transfers one way and the other.Heat cannot spontaneously flow from cold regions to hot regions without external work being performed on the system, which is evident from ordinary experience of refrigeration, for example. In a refrigerator, heat flows from cold to hot, but only when forced by an external agent, the refrigeration system.
Kelvin statement
- It is impossible, by means of inanimate material agency, to derive mechanical effect from any portion of matter by cooling it below the temperature of the coldest of the surrounding objects
Equivalence of the Clausius and the Kelvin statements
- Suppose there is an engine violating the Kelvin statement: i.e., one that drains heat and converts it completely into work in a cyclic fashion without any other result. Now pair it with a reversed Carnot engine as shown by the graph. The net and sole effect of this newly created engine consisting of the two engines mentioned is transferring heat
 from the cooler reservoir to the hotter one, which violates the Clausius statement. Thus a violation of the Kelvin statement implies a violation of the Clausius statement, i.e. the Clausius statement implies the Kelvin statement. We can prove in a similar manner that the Kelvin statement implies the Clausius statement, and hence the two are equivalent.
from the cooler reservoir to the hotter one, which violates the Clausius statement. Thus a violation of the Kelvin statement implies a violation of the Clausius statement, i.e. the Clausius statement implies the Kelvin statement. We can prove in a similar manner that the Kelvin statement implies the Clausius statement, and hence the two are equivalent. Clausius Inequality
- The Clausius Theorem states that in a cyclic process
-

Entropy
- According to the Clausius equality, for a reversible processThat means the line integral
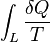 is path independent.So we can define a state function S called entropy, which satisfies
is path independent.So we can define a state function S called entropy, which satisfies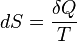
- With this we can only obtain the difference of entropy by integrating the above formula. To obtain the absolute value, we need the Third Law of Thermodynamics, which states that S=0 at absolute zero for perfect crystals.For any irreversible process, since entropy is a state function, we can always connect the initial and terminal status with an imaginary reversible process and integrating on that path to calculate the difference in entropy.Now reverse the reversible process and combine it with the said irreversible process. Applying Clausius inequality on this loop,Thus,where the equality holds if the transformation is reversible.
Derivation of the entropy change for reversible processes
- The second part of the Second Law states that the entropy change of a system undergoing a reversible process is given by:where the temperature is defined as:See here for the justification for this definition. Suppose that the system has some external parameter, x, that can be changed. In general, the energy eigenstates of the system will depend on x. According to the adiabatic theorem of quantum mechanics, in the limit of an infinitely slow change of the system's Hamiltonian, the system will stay in the same energy eigenstate and thus change its energy according to the change in energy of the energy eigenstate it is in.The generalized force, X, corresponding to the external variable x is defined such that
 is the work performed by the system if x is increased by an amount dx. E.g., if x is the volume, then X is the pressure. The generalized force for a system known to be in energy eigenstate
is the work performed by the system if x is increased by an amount dx. E.g., if x is the volume, then X is the pressure. The generalized force for a system known to be in energy eigenstate  is given by:Since the system can be in any energy eigenstate within an interval of
is given by:Since the system can be in any energy eigenstate within an interval of , we define the generalized force for the system as the expectation value of the above expression:To evaluate the average, we partition the
, we define the generalized force for the system as the expectation value of the above expression:To evaluate the average, we partition the energy eigenstates by counting how many of them have a value for
energy eigenstates by counting how many of them have a value for 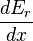 within a range between
within a range between 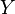 and
and  . Calling this number
. Calling this number  , we have:The average defining the generalized force can now be written:We can relate this to the derivative of the entropy w.r.t. x at constant energy E as follows. Suppose we change x to x + dx. Then will change because the energy eigenstates depend on x, causing energy eigenstates to move into or out of the range between
, we have:The average defining the generalized force can now be written:We can relate this to the derivative of the entropy w.r.t. x at constant energy E as follows. Suppose we change x to x + dx. Then will change because the energy eigenstates depend on x, causing energy eigenstates to move into or out of the range between 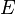 and
and 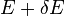 . Let's focus again on the energy eigenstates for which lies within the range between and . Since these energy eigenstates increase in energy by Y dx, all such energy eigenstates that are in the interval ranging from E – Y dx to E move from below E to above E. There aresuch energy eigenstates. If
. Let's focus again on the energy eigenstates for which lies within the range between and . Since these energy eigenstates increase in energy by Y dx, all such energy eigenstates that are in the interval ranging from E – Y dx to E move from below E to above E. There aresuch energy eigenstates. If , all these energy eigenstates will move into the range between and and contribute to an increase in
, all these energy eigenstates will move into the range between and and contribute to an increase in 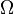 . The number of energy eigenstates that move from below to above is, of course, given by
. The number of energy eigenstates that move from below to above is, of course, given by  . The differenceis thus the net contribution to the increase in. Note that if Y dx is larger than there will be the energy eigenstates that move from below E to above . They are counted in both
. The differenceis thus the net contribution to the increase in. Note that if Y dx is larger than there will be the energy eigenstates that move from below E to above . They are counted in both  and , therefore the above expression is also valid in that case.Expressing the above expression as a derivative w.r.t. E and summing over Y yields the expression:The logarithmic derivative of w.r.t. x is thus given by:The first term is intensive, i.e. it does not scale with system size. In contrast, the last term scales as the inverse system size and will thus vanishes in the thermodynamic limit. We have thus found that:Combining this withGives:
and , therefore the above expression is also valid in that case.Expressing the above expression as a derivative w.r.t. E and summing over Y yields the expression:The logarithmic derivative of w.r.t. x is thus given by:The first term is intensive, i.e. it does not scale with system size. In contrast, the last term scales as the inverse system size and will thus vanishes in the thermodynamic limit. We have thus found that:Combining this withGives: Third law
- The third law of thermodynamics is sometimes stated as follows, regarding the properties of systems in equilibrium at absolute zero temperature:
- The entropy of a perfect crystal, at absolute zero (zero kelvins), is exactly equal to zero.
- At zero kelvin the system must be in a state with the minimum possible energy, and this statement of the third law holds true if the perfect crystal has only one minimum energy state. Entropy is related to the number of possible microstates, and for a system containing a certain collection of particles, quantum mechanics indicates that there is only one unique state (called the ground state) with minimum energy
Mathematical formulation
- Consider a closed system in internal equilibrium. As the system is in equilibrium there are no irreversible processes so the entropy production is zero. During the heat, supply temperature gradients are generated in the material, but the associated entropy production can be kept low enough if the heat is supplied slowly. The increase in entropy due to the added heat δQ is then given by the second part of the Second law of thermodynamics which states that the entropy change of a system is given by

(1) The temperature rise dT due to the heat δQ is determined by the heat capacity C(T,X) according to
(2) The parameter X is a symbolic notation for all parameters (such as pressure, magnetic field, liquid/solid fraction, etc.) which are kept constant during the heat supply. E.g. if the volume is constant we get the heat capacity at constant volume CV. In the case of a phase transition from liquid to solid, or from gas to liquid the parameter X can be one of the two components. Combining relations (1) and (2) gives
(3) Integration of Eq.(3) from a reference temperature T0 to an arbitrary temperature T gives the entropy at temperature T
(4) We now come to the mathematical formulation of the third law. There are three steps:1: in the limit T0→0 the integral in Eq.(4) is finite. So that we may take T0=0 and write
(5) 2. the value of S(0,X) is independent of X. In mathematical form
(6) So Eq.(5) can be further simplified to
(7) Equation (6) can also be formulated as
(8) In words: at absolute zero all isothermal processes are isentropic. Eq.(8) is the mathematical formulation of the third law.3: as one is free to chose the zero of the entropy it is convenient to take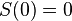
(9) so that Eq.(7) reduces to the final form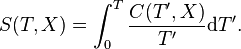
(10)




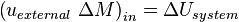










 , so
, so  .
.
![\frac{1}{k_{\mathrm B} T}\equiv\beta\equiv\frac{d\ln\left[\Omega\left(E\right)\right]}{dE}](http://upload.wikimedia.org/math/9/a/a/9aa130c8295d811f02f994634ee80ca4.png)











No comments:
Post a Comment